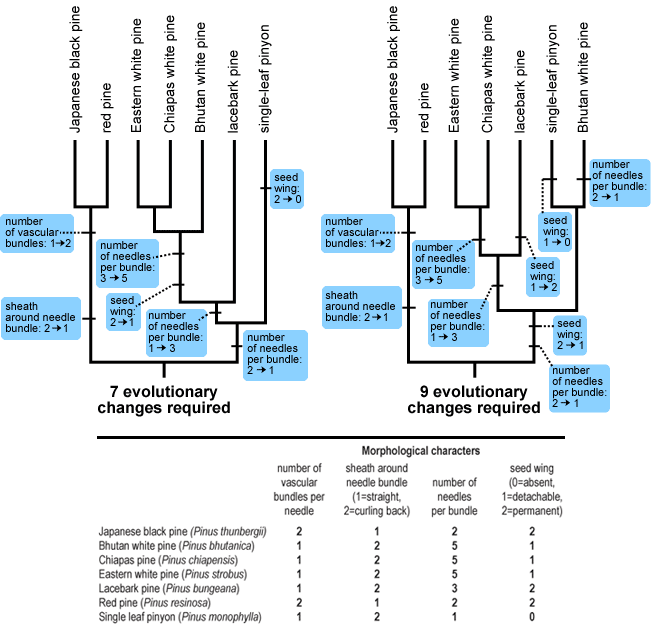
One reliable method of building and evaluating trees, called parsimony, involves grouping taxa together in ways that minimize the number of evolutionary changes that had to have occurred in the characters. The idea here is that, all other things being equal, a simple hypothesis (e.g., just four evolutionary changes) is more likely to be true than a more complex hypothesis (e.g., 15 evolutionary changes). So, for example, based on the morphological data, the tree at left below requires only seven evolutionary changes and, based on the available evidence, is a better hypothesis than the tree at right, which requires nine evolutionary changes.
To find the tree that is most parsimonious, biologists use brute computational force. The idea is to build all possible trees for the selected taxa, map the characters onto the trees, and select the tree with the fewest number of evolutionary changes. It’s a simple idea, but the first two steps require a lot of work — or a lot of computing power!
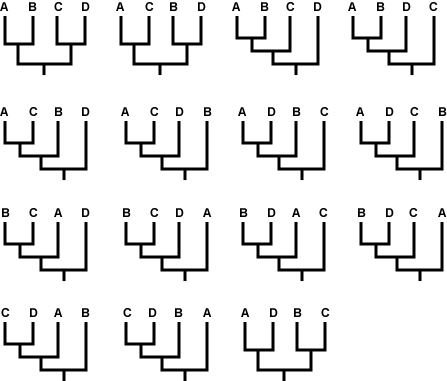
First, what is meant by “build all possible trees?” Imagine that we want to figure out the evolutionary relationships among just four taxa: A, B, C, and D. There are 15 different ways that those taxa could be related, shown below, and that number skyrockets as the number of taxa increases. For just 10 taxa, there are more than 34 million different possible trees! So the first step to building a tree using parsimony is not trivial. Because of the huge number of possible trees — far too many to be dealt with on paper — biologists use computer programs designed for this task.
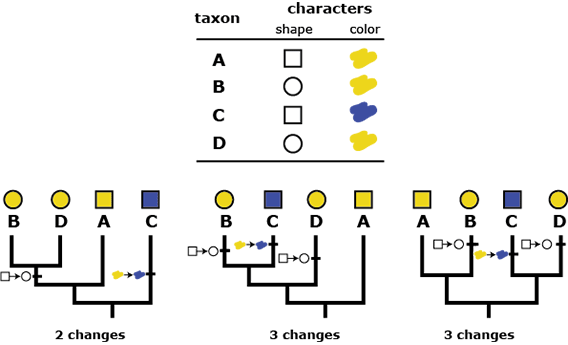
Next, evolutionary transitions in each character are parsimoniously mapped onto each of the possible trees, and biologists select the tree that requires the fewest number of evolutionary changes. So for example, consider just three of the phylogenies shown above and a data matrix of two characters. The character data are mapped onto each tree in the most parsimonious way possible, but one of the trees is clearly more parsimonious than the others. Tree 1 requires just two changes in characters to account for the data, while Trees 2 and 3 require three changes to account for the data. If we were biologists using parsimony to select among these three trees, we would select the leftmost tree below as the most likely to be accurate because it hypothesizes the simplest evolutionary trajectory that accounts for the evidence we’ve collected. (Note that for the example above with four taxa and 15 possible trees, there are multiple character reconstructions on different trees that result in just two evolutionary changes, not just the leftmost tree shown above. In practice, biologists use many more than two characters to evaluate trees, and outgroups are used to constrain likely ancestral character states, resulting in fewer “ties” for most parsimonious tree.)
It’s easy to see how complex this process could become with a large number of taxa and characters. Biologists often use data matrices with tens or hundreds of taxa and thousands of characters. Computer programs help them keep track of the huge number of possible trees and all the different ways that the character data could be mapped onto each tree.
Ideally, this process of hunting through all possible would result in a single, perfect tree. But of course, evolution is not a directed, linear process and so sometimes doesn’t actually happen in the most parsimonious way. Sometimes similar changes occur in different lineages and we just can’t tell that convergence has occurred. And sometimes evolution backtracks and a trait that arose in a lineage disappears or returns to its prior state. For these reasons, in practice some characters are likely to support one tree shape, and others are likely to support different tree shapes. This is why it’s important to collect evidence from many different characters — so that we can be confident which tree the bulk of the evidence supports. When conflicts like these arise, the best-supported tree is the one that is the most parsimonious (i.e., requires the fewest evolutionary changes).
Feeling lost? Review tree basics with the primer.